
Once in a while you see an idea that is both so brilliant and so obvious that you just have to smack your head and ask “Why has no one thought of this before? Why didn’t I think of it?”.
There’s a general consensus that broad-spectrum antibiotics are over-used. The consequences are many and egregious: disruption of healthy microbiomes, leading to increased risk of serious diseases like C difficile diarrhea, and possibly of auto-immune and metabolic diseases; encouragement of the spread of resistance between bacterial strains.
Not a few expert clinicians have argued for the development of narrow-spectrum agents that would go easy on the microbiome and reduce selection pressure for resistance.
We’ve made very slight progress. Fidaxomicin, which selectively targets C. difficile, has been approved and seems to result in lower relapse rates for serious diarrhea than broad-spectrum agents like vancomycin and metronidazole.
But narrow-spectrum agents have not met much commercial success. Sales of fidaxomicin appear stuck at ~$60M/year (any sales number not denominated with a “B” is considered boring by investors). Achaogen was developing a solid pipeline of narrow-spectrum agents; it recently filed for bankruptcy.
The problem with narrow-spectrum agents, from a revenue standpoint, is that they by definition have narrow markets. Worse, they require a micro work up in order to ID the bug and justify their prescription. If there is anything that the marketers who run drug companies hate more than small markets and companion diagnostics, I don’t know what it is. Given their high societal utility but low potential for profit, I think narrow-spectrum antibiotic development is a clear instance of market failure and should be socialized.
Until that happens, though, we may need to rely on technology combined with some creative thinking. A group of medicinal chemists at Imperial College in London had this idea: why not create a pro-drug antibiotic that is activated by drug resistance enzymes? In other words, create a Trojan Horse antibiotic that targets only resistant strains and leaves susceptible strains alone.
Many of the most troublesome forms of antibiotic resistance are mediated by enzymes that cleave antibiotics and render them harmless. Resistance to cephalosporins and carbapenems, critical for treating Gram-negative infections such as E coli and Klebsiella, fall into this category.
The strategy was to hook a cephalosporin (a first-line antibiotic) on to a second-line antibiotic (ciprofloxacin). The conjugated antibiotic is 10-30 fold less potent than either antibiotic alone, meaning that it should have no effect on bacteria susceptible to cephalosporins. Cephalosporin-resistant bacteria, however, will cleave off the cephalosporin group, activating the ciprofloxacin portion:
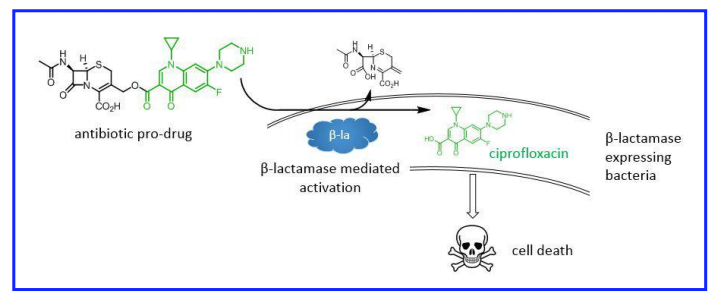
In principle, only the bad bugs–the ones that express β-lactamases–are killed by ciprofloxacin. Bugs that are susceptible to treatment with first-line β-lactam drugs (cephalosporins) are left alone. As a bonus, systemic levels of ciprofloxacin, which has significant side-effects, are kept low, reducing the risk of toxicity.
That’s the principle, how does it work in practice?

The pro-drug is about 10-fold less active than naked ciprofloxacin against strains that have no β-lactamase-mediated resistance to cephalosporins and carbapenems (WT and pEMP above, blue lines shifted to right). That gap narrows to about 2-fold in strains that bear notoriously problematic β-lactamases (CTX, NDM1 and KPC).
Susceptible strains grow just about as well when treated with the pro-drug as they do with no drug:

Only the cephalosporin-resistant strains are killed. The upshot is that the selective pressure to acquire resistance is reduced and inverted: the most straightforward way to become resistant to the pro-drug is to lose the B-lactamase enzyme and become susceptible to cephalosporins. It’s a form of microbial jiu-jitsu.
This is still all a long way from the clinic. We don’t know anything about the pharmacology or toxicity of this particular pro-drug. But the concept is sound, it’s just a matter of optimizing the chemistry. That will be a lot of work, but it’s just work. The breakthrough has been made.
The brilliance of this idea is that it redefines the concept of narrow-spectrum antibiotics. Rather than targeting a narrow range of species, this class of drugs narrows its targets by their resistance phenotypes. And because the drug is only activated within and near its targets, it is also narrowed spatially, reducing the risk of systemic side-effects.
Perhaps most importantly, these drugs might prove economically attractive. One could imagine physicians prescribing them with no micro workup, and perhaps even combined with cephalosporins as a first-line treatment. It’s possible, just possible, that the code has been cracked to make antibiotic development economically attractive again. That’s a huge deal.