
One can sum up the 100-year history of phage therapy concisely: works great in the lab, fails in the clinic. Not a single Phase 2 or 3 clinical trial has yet shown phage therapy to be effective.
This failure is not due to a lack of research effort. Search for “phage therapy” in PubMed and you’ll get over 3200 hits, the majority of those being published in the 21st century.
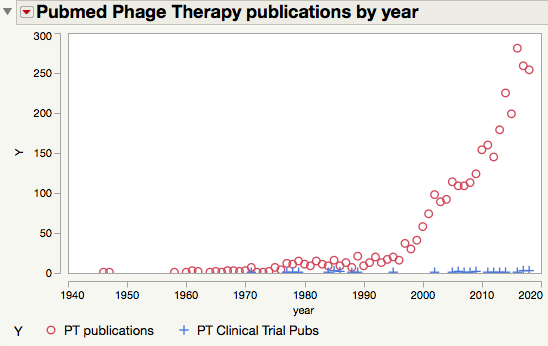
But motion is not the same as progress. Progress requires a willingness to fail, and the foresight to design your experiments so that failures are informative. You want to be able to say, “Well, that didn’t work, but we know why. Let’s try this instead”. What you really don’t want to do is structure your studies so that they are a success–meaning that they confirm your hypothesis–but can’t plausibly be translated into human therapies. That is an empty and meaningless definition of success. The point of biomedical research is to benefit your fellow humans, not to rack up publications.
Developing therapeutics is hard. By one estimate, only about 4% of leads make it from preclinical studies all the way into clinical use. Most folks who think about this will tell you that the main cause of failure is that preclinical animal models don’t accurately predict therapeutic responses in humans. We pretty much know how to cure cancer in mice (I have saved a few rodents from cancer myself), we just can’t do it in humans.
But antimicrobial therapy is not as hard a problem. After all, the disease-causing entity is not intrinsic, like Alzheimer’s or cancer, but extrinsic. Things that kill bugs in a test tube will also kill them in patients. All you have to do is (a) make sure they are not toxic and (b) make sure the drug will reach its target in sufficient concentration.
Those aren’t trivial challenges, but they lower the bar quite a bit. And indeed, anti-infectives have likelihoods of approval that are among the highest of any class of drugs:
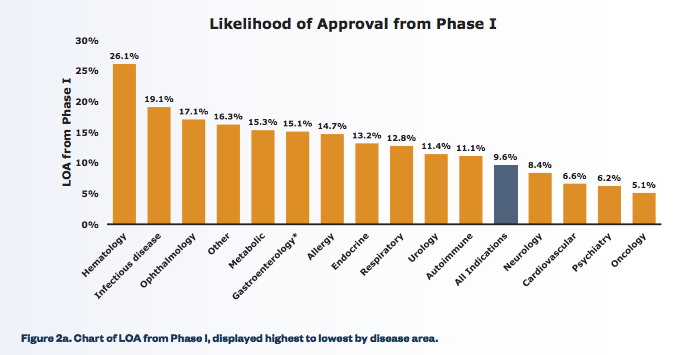
From Clinical Development Success Rates, 2006–2011
No phage therapy trial has failed because of safety issues, which is the downfall of many new molecular entities. Phage are in fact classified as “Generally Recognized As Safe” by the FDA, and phage therapy studies can largely bypass costly ADME studies in order to get the go-ahead (an IND) for conducting clinical trials.
To sum up:
- Numerous studies show the efficacy of phage therapy in preclinical testing.
- Phage therapy is a member of a class of therapies that have high success rates in clinical trials.
- Phage are unlikely to fail trials due to safety concerns,.
- And yet, phage have never succeeded in a late-stage clinical trial.
Why?
Here’s my hypothesis: most of the preclinical studies of PT in animal models are pure bullshit. They are intended to get a cheap and easy publication, and maybe a press release hailing this “new old therapy” or similar bunk.
Let’s take a look at the latest example that has landed in my “Phage therapy” alert feed in PubMed, titled “Form[ul]ation of therapeutic phage cocktail and endolysin to highly multi-drug resistant Acinetobacter baumannii: in vitro and in vivo study“.
The authors are tackling a serious problem —Acinetobacter infections are notoriously resistant to antibiotic therapy, and are especially prevalent in the Middle East (the study was done in Iraq). A case study in which phage therapy appeared to cure a near-fatal antibiotic resistant Acinetobacter infection gained quite a bit of notoriety in the US (I dumped on it here).
The authors did a nice job of isolating and characterizing the phage. They then showed that the phage could protect mice against Acinetobacter bloodstream infections. The basic procedure was to inject the mice with a high dose of bacteria, and then follow that with high doses of phage. This saved the mice. Unfortunately, the chance that these results have any clinical relevance is approximately zero. Here are the problems:
- Bacteremia is a secondary infection. There is always a primary source (pneumonia, a wound, a urinary tract infection) from which bacteria escape into the blood. This is bad because it allows the infection to metastasize, and because it increases the likelihood that the immune system will over-react, leading to septic shock. In actual patients, clearing bacteria from the blood but not the primary locus is unlikely to have much clinical benefit.
- The concentrations of both bacteria and phage are unrealistically high. Most human bloodstream infections have bacterial concentrations of ≤1 cell per mL. 100 per mL would be very high, and death imminent. The study protocol called for 5 x 105 bacteria to be injected. Mice typically have a plasma volume of 1.5 mL, so this works out to about 3 x 105 per mL, thousands of times more bacteria than encountered in a human case of bacteremia.
- High doses of bacteria are required to see a clinical response in mice – rodents are much more tolerant than humans of bacteria in the blood. This is a problem in using mouse models of bacteremia. But using high doses also makes experimentation much easier – the mice sicken and die within hours, something that rarely happens in human bacterial infections.
- The phage were given at a dose 3 x 107, for an initial concentration of about 2 x 107 per mL. This is higher than the initial concentrations reported in recent case studies (here and here) by a factor of well over a hundred. Systemic phage therapy requires these high doses, but no one is achieving them, largely due to inability to purify phage away sufficiently from endotoxin. If this is indeed a fundamental limit in phage therapy doses, that limit needs to be incorporated into experimental design.
- With bacteria and phage at such high concentrations, the mice acted essentially as animate test tubes. It’s not a surprise that the phage attacked the bacteria quickly and efficiently and rescued the mice from otherwise fatal doses of bacteria.
- Phage were given 2 hours after bacterial infection. This is highly unlikely in any clinical scenario. Other studies of phage therapy in animal models show significant dropoffs in efficacy after more than a few hours (see here and here). Curing established infections is obviously much harder. But it is also the only realistic model.
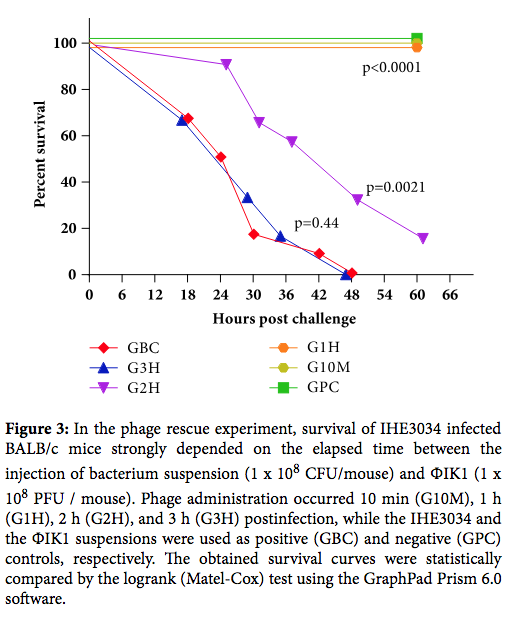
From Kinetics of Targeted Phage Rescue in a Mouse Model of Systemic Escherichia coli K1
The Iraqi paper is hardly the worst phage therapy study I’ve seen; in fact, it is probably one of the better ones. But that is faint praise. It’s almost 2019, and the animal models we are using for preclinical studies of phage therapy are no better than those of a decade or two ago. They are laughably weak and unrealistic and completely unlikely to uncover the problems that will arise in the clinic.
The point of using models is to reveal problems so that you can fix them. It is not to cure mice of infections. Until the phage therapy community demands more-realistic preclinical models, ones that fail in ways that human studies are likely to fail, we are never going to make progress. That’s not okay.
Hi Drew, thanks for this great post. What model would you propose instead? Would it be better to infect the mice at lower bacterial load and wait longer? Different animal altogether?
I’m not much of an animal physiologist, so I can’t give a specific suggestion. But here are the features I’d like to see in an animal model of bacteremia: 1. That the bacteremia be a secondary manifestation of a primary infection, such as pneumonia, a wound, organ, UTI etc. 2. That phage therapy be shown effective against established infections over a wide range of times post-infection. The obvious comparator here is antibiotic therapy. If a PT regimen is not comparable to antibiotics, it probably doesn’t have much clinical utility. It doesn’t have to be better, just comparable. 3. The doses have to be scalable to human dosing. Realistically I think that means several different models, which is a lot of work. But showing efficacy in several models would give real confidence that preclinical results would translate to clinical utility. 4. Better purification schemes to get rid of endotoxin, which has been dose-limiting. That is not so hard, and it is really shameful that it has not been done already.
Thank you for the comments. It makes a lot of sense to use phage against bacteremia that is as a result of something else. I wonder if it would be extremely difficult to control for. The time to bacteremia from wound infection could vary drastically from one mouse to another.